Combined or hybrid Quantum Mechanics and Molecular Mechanics (QM/MM) is a simulation methodology 33.1 that is about 15 years old but in all the literature there are cautions that calibration computations must be done to validate the model for each particular chemical system studied. This is not a black box style computation and the NWChem users are advised that without calibration QM/MM may not give the appropriate results. Since both quantum-mechanical and classical molecular mechanics are involved in the calculation good working knowledge of the two methods is required to ensure meaningful results.
The QM/MM module is invoked with the following task directive.
task qmmm <string qmtheory> <string operation> [numerical] [ignore]where qmtheory specifies quantum method for the calculation of the quantum region. It is expected that most of the QM/MM simulations will be performed with HF or DFT theories, but any other QM theory supported by NWChem should work as well. Currently the supported operations for QM/MM runs are energy, optimize, saddle, dynamics, numerical hessian, and numerical frequencies.
Unlike pure quantum mechanical calculations the information about the chemical system for QM/MM simulations is contained not in the geometry block but in the externally prepared topology and restart files. These files have to be present prior to any QM/MM simulation. The input file for QM/MM simulation can be divided into three major parts - specification of the molecular mechanics parameters for the classical region, specification of the quantum mechanical method for the quantum region, and the parameters of the interaction between quantum and classical methods. All this discussed in detail in the sections below.
Generated by the prepare module (see section 30) restart and topology files contain information about the classical force field as well as the coordinates of quantum (qm) and molecular mechanics (mm) regions. In a typical setting this "preparation stage" will be run separately from main QM/MM simulation. This will require a properly formatted PDB file for the system. In more complex cases (e.g.non-standard residues or nucleotides) additional fragment and parameter files might have to be provided by the user. The definition of the quantum region in the input for the prepare module is specified by either modify atom directive (see Section 30):
modify atom <integer isgm>:<string atomname> quantum
or modify segment directive
modify segment <integer isgm> quantum
Here isgm and atomname refer to the residue number and atom name record as given in the PDB file. It is important to note that that the leading blanks in atom name record should be indicated with underscores. Per PDB format quidelines the atom name record starts at column 13. If, for example, the atom name record "OW" starts in the 14th column in PDB file, it will appear as "_OW" in the modify atom directive in the prepare block. In the current implementation only solute atoms can be declared as quantum. If part of the solvent has to be treated quantum mechanically then it has to redeclared to be solute. In addition to modify commands the prepare input block should also contain update lists and ignore directives. There are other options that can be used in the input block for the prepare module ( e.g. solvating the structure, etc ), those discussed in more details in Section 30. The successful run of the prepare module will result in generation of topology and restart files. Similar to classical MD, both files are required for QM/MM simulations and have to be placed in the same directory as the input file. Here is an example input file that will generate QM/MM restart and topology files for the ethanol molecule
title "Prepare QM/MM calculation of ethanol" start etl prepare #--name of the pdb file source etl0.pdb #--generate new topology and sequence file new_top new_seq #--generate new restart file new_rst #--define quantum region (note the use of underscore) modify atom 1:_C1 quantum modify atom 1:2H1 quantum modify atom 1:3H1 quantum modify atom 1:4H1 quantum # update lists ignore end task prepare
These are contents of etl0.pdb file used in the above input file.
ATOM 1 O etl 1 1.201 -0.271 -0.000 1.00 0.00 O ATOM 2 H etl 1 1.995 0.329 -0.000 1.00 0.00 H ATOM 3 C1 etl 1 -1.180 -0.393 0.000 1.00 0.00 C ATOM 4 2H1 etl 1 -2.128 0.155 -0.000 1.00 0.00 H ATOM 5 3H1 etl 1 -1.130 -1.030 0.887 1.00 0.00 H ATOM 6 4H1 etl 1 -1.130 -1.030 -0.887 1.00 0.00 H ATOM 7 C2 etl 1 0.006 0.573 0.000 1.00 0.00 C ATOM 8 2H2 etl 1 -0.042 1.220 0.890 1.00 0.00 H ATOM 9 3H2 etl 1 -0.042 1.220 -0.890 1.00 0.00 H END
Running the input shown above will produce (among other things) the topology file (etl.top) and the restart file (etl_md.rst). The naming of the topology file follows after the rtdb name specified in the start directive in the input (i.e. "start etl"), while the "_md" suffix in the restart file name is specific to the way prepare module works in this particular case. If necessary, this particular naming scheme can be altered using system keyword in the prepare input block (for more details see Section 30).
md # this specifies that etl_md.rst will be used as a restart file # and etl.top will be a topology file system etl_md # if we ever wanted to fix C1 atom fix solute 1 _C1 end
The parameters defining calculation of the QM region (including basis sets) must be present in the traditional NWChem input format except for the geometry block. The geometrical information will be constructed automatically by QM/MM using information from the MD module.
The QM/MM interface parameters define the interaction between classical and quantum regions. The input follows standard NWChem format:
qmmm
[ eref <double precision default 0.0d0>]
[ bqzone <double precision default 9.0d0>]
[ bq_exclude <(none||all||linkbond||linkbond_H) default none>]
[ bq_update <(static||dynamic) >]
[ link_atoms <(hydrogen||halogen) default hydrogen>]
[ link_ecp <(auto||user) default auto>]
[ optimization <(all||mm|qm) default qm>]
end
Detailed explanation of the subdirectives in the QM/MM input block is given below:
eref <double precision default 0.0d0>
This directive sets the relative zero of energy for the QM component of the system. The need for this directive arizes from different definitions of zero energy for QM and MM methods. Most QM methods define the zero of energy for the system as vacuum. The zero of energy for the MM system is by definition of most parameterized force fields the separated atom energy. Therefore in many cases the energetics of the QM system will likely overshadow the MM component of the system. This imbalance can be corrected by suitably chosen value of eref
bqzone <double precision default 9.0d0>
This directive defines the radius of the zone (in angstroms) around the quantum region
where classical residues/segments
will be allowed to interact with quantum region electrostatically. It should be noted
that classical atoms interacting with quantum region via bonded interactions are always
included in the bqzone (this is true even if bqzone is set to ![]() ). In addition, even if one atom
of a given charged group is in the bqzone (residues are typically treated as one charged group) then the whole
group will be included in the bqzone.
). In addition, even if one atom
of a given charged group is in the bqzone (residues are typically treated as one charged group) then the whole
group will be included in the bqzone.
bq_exclude <(none||all||linkbond||linkbond_H) default none>
This directive operates in conjunction with bqzone keyword offering additional level of control on electrostatic interactions between quantum and classical regions. Default value none will leave bqzone unchanged, all will remove all electrostatic interactions between classical and quantum regions, linkbond will result in the removal of electrostatic interactions with atoms that are connected to a quantum region by at most two bonds, linkbond_H is similar to linkbond but will only remove interactions with hydrogen atoms. If necccessary, further modifications of the electrostatic interactions can be achieved using the prepare module30.
bq_update <(static||dynamic)This directive controls whether the bqzone stays fixed (static) or constantly updated (dynamic) during the calculation. In most cases direct specification of this keyword will not be necessary as its value will be set automatically based on the nature of the calculation.
link_atoms <(hydrogen||halogen) default halogen>
This directive controls the treatment of bonds crossing the boundary between quantum and classical regions.
The use of hydrogen keyword will trigger truncation of such bonds with hydrogen link atoms. The position of the hydrogen
atom will be calculated from the coordinates of the quantum and classical atom of the truncated bond using the
following expression
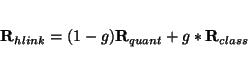
Setting link_atoms to halogen will result in the modification of the quantum atom of the truncated bond to to the fluoride atom. This fluoride atom will typically carry an effective core potential (ECP) basis set as specified in link_ecp directive.
link_ecp <(auto||user) default auto>This directive specifies ECP basis set on fluoride link atoms. If set to auto the ECP basis set given by Zhang, Lee, Yang for 6-31G* basis.33.2 will be used. Strictly speaking, this implies the use of 6-31G* spherical basis as the main basis set. If other choices are desired then keyword user should be used and ECP basis set should be entered separatelly following the format given in section 8. The name tag for fluoride link atoms is F_L.
optimization <(all||mm|qm) default qm>
This directive specifies which region will be optimized during QM/MM optimization. If set to all both qm and mm region will be optimized simultaneously using mm optimization module, if mm only mm portion will be optimized using mm optimization module, finally if qm is specified then only qm region will be optimized using driver module. Since option all is based on steepest descent minimization algorithm it is not reccomended for accurate QM/MM optimization tasks. The most efficient and accurate way to perform QM/MM optimizations is to optimize quantum region using optimization qm followed by the relaxation of the mm region using optimization mm.